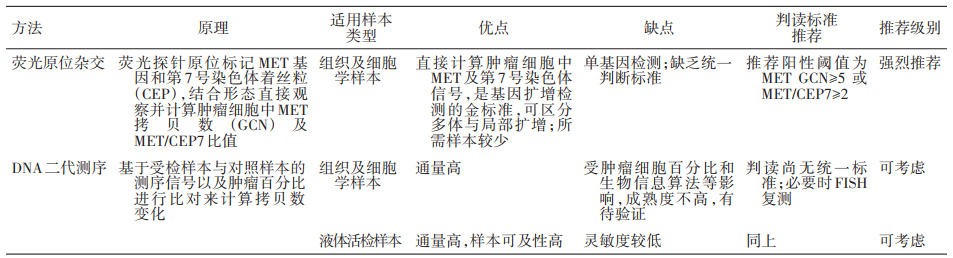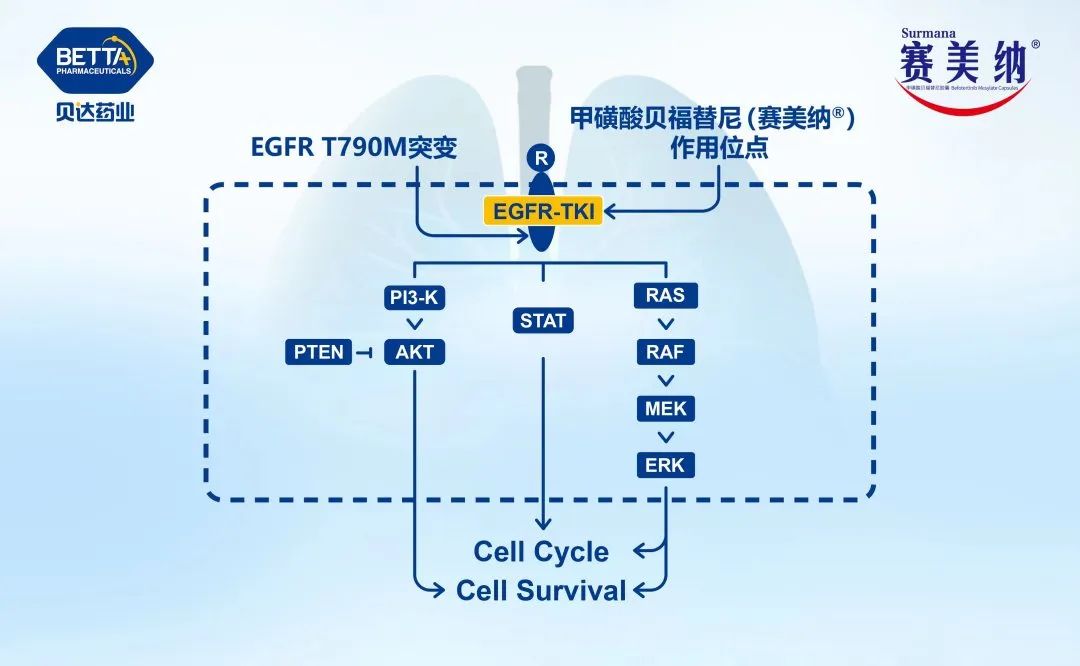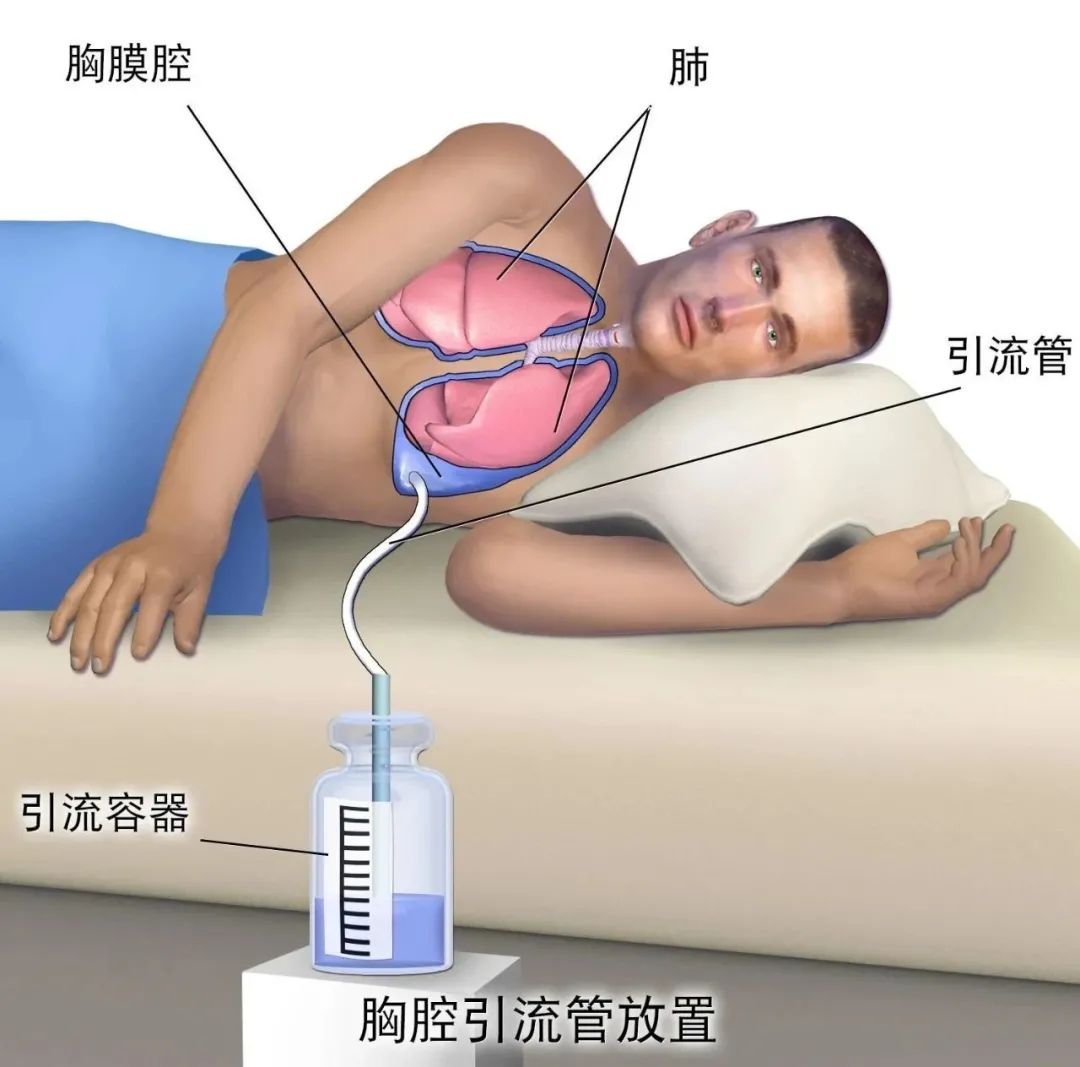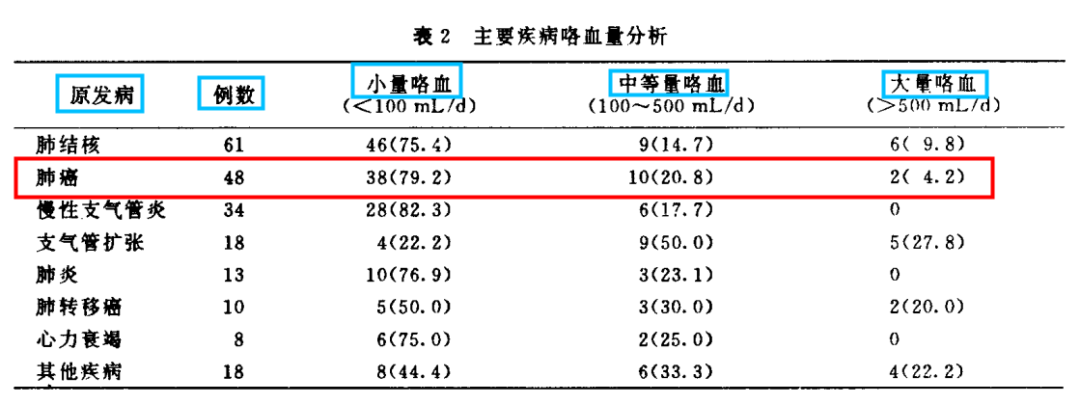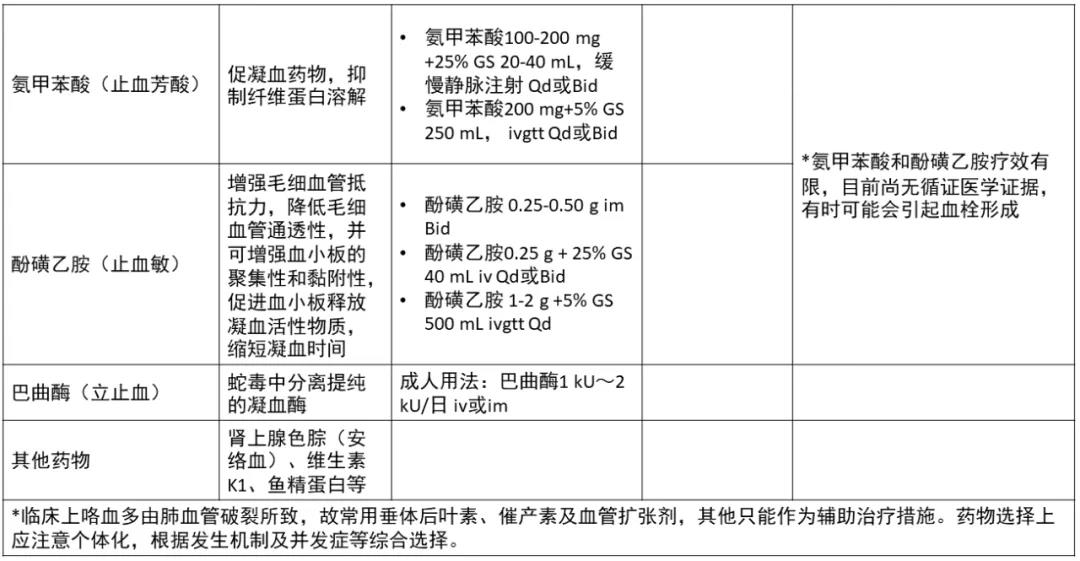

表皮生长因子受体(EGFR)是一种跨膜受体,是酪氨酸激酶受体家族成员之一,目前针对EGFR为靶点的药物主要分为两大类:临床最常见药物制剂是酪氨酸激酶抑制抑制剂(TKI),即表皮生长因子受体酪氨酸激酶抑制抑制剂(EGFR-TKI);一类是临床少见的针对EGFR的单克隆抗体。诸多前瞻性临床研究均证实第一代EGFR-TKI治疗EGFR敏感突变的晚期非小细胞肺癌(NSCLC)在无疾病进展时间(PFS)和客观缓解率(ORR)方面明显优于传统的含铂两药联合方案,充分奠定了EGFR-TKI在EGFR敏感突变患者的一线治疗地位。本文将对EGFR-TKI药物在临床治疗NSCLC方面的研究进行归纳论述,以期为临床合理选择EGFR-TKI提供参考。 第一代口服EGFR-TKI的代表药物如吉非替尼、厄洛替尼和埃克替尼,虽然在初治的携带药物敏感EGFR基因突变的NSCLC患者获得良好的疗效,并且一代EGFR-TKI较化疗一线药物,可改善其无进展生存期,但是随着用药时间的延长会出现不同程度的耐药,一般治疗7~14个月后出现EGFR-TKI耐药,因此,临床着重研究EGFR-TKI耐药机制及后续的进一步诊治是有待解决的问题。 原发性耐药是指首次使用EGFR-TKI治疗无反应,关于EGFR原发性耐药的原因有很多,其主要原因包括以下几个方面:(1)存在与药敏突变不同的其他EGFR突变点,除外19del和21L858R两种常见的敏感突变,20ins或其他的罕见突变可能与EGFR-TKI耐药有关;(2)K-RAS基因突变。研究结果表明,在TKI不敏感的患者中有大约25%左右的肿瘤组织中存在着K-RAS基因突变,且另一项随机对照研究也表明了KRAS基因突变是影响TKI药物疗效的因素之一;(3)患者自身的因素。如新陈代谢的快速失活,免疫功能低下,以及吸收能力的降低等。 获得性耐药定义:(1)既往接受过EGFR-TKI单药治疗;(2)符合以下条件之一:存在EGFR-TKI基因敏感突变(如G719X、19del、L858R和L861Q);一线使用EGFR-TKI有临床获益,包括完全缓解、部分缓解或大于6个月的疾病稳定;(3)持续接受EGFR-TKI治疗至少一个月后疾病进展;停用EGFR-TKI及开始新的治疗前未接受其他全身治疗。虽然在EGFR突变的NSCLC中一代EGFR-TKI药物可以起到不错的疗效,但大部分患者在治疗7~11个月后会出现不同程度的获得性耐药。目前关于EGFR-TKI获得性耐药的机制主要有以如下几个方面: 2.2.1 EGFR二次点突变 目前最普遍认同的是二次点突变学说,主要是T790M突变,此类患者多见于不吸烟的肺腺癌患者,也存在其他少见的突变如:T854A及D761Y突变等。研究发现T790M是由EGFR相关基因的缺失引起突变。此二次突变机制常见的原因是患者在服用一代EGFR-TKI药物(如:厄洛替尼或吉非替尼)治疗后,EGFR基因在原有突变的基础上产生了新的突变,即EGFR基因20号外显子第790号位点出现了C与T交换的现象氨基酸发生改变,产生位阻现象,使EGFR与一代EGFR-TKI药物之间无法正常结合,然而EGFR与ATP之间的结合没有明显变化,对相关激酶磷酸化的影响也有限,可引起其下游相关信号通路的变化,从而产生了获得性耐药。 2.2.2 c-MET扩增 c-MET是肝细胞生长因子(HGF)受体酪氨酸激酶,在胚胎发育、组织修复和肿瘤进展中起关键作用,在胃癌和NSCLC等癌症的不同阶段,甚至在使用EGFR-TKI治疗后,也经常观察到c-MET的过表达和扩增。c-MET引起耐药机制的方式很多,其突变能够促进血管生成,参与上皮活动,促进肿瘤生长也能够编码EGFR并通过激活P13K/AKT信号通路,进一步引起EGFR-TKI耐药,它的突变占第一代EGFR-TKI耐药机制的20%左右。研究报道通过第一代EGFR-TKI药物吉非替尼诱导HCC827细胞株后产生耐药并检测到c-MET扩增,在应用c-MET相应通路抑制剂后发现能够使耐药细胞株表现出对一代EGFR-TKI药物的敏感性。 2.2.3 HER2突变 NSCLC中约2%~4%的病例发生HER2突变,最常见于从不吸烟的肺腺癌组织学中。最常见的突变由外显子20中的12个碱基对插入或重复,如:YVMA的插入。同样在非小细胞肺癌中,HER2的致癌扩增发生在未经EGFR-TKI治疗的病例中约3%,在EGFR-TKI耐药性中约占病例的10%。HER2扩增和突变通常不会同时发生。肺癌中HER2激活的第三个机制是蛋白质过表达,据报道在2%~20%的病例中会发生这种情况,具体取决于其过表达的水平(IHC 2+或3+),并显示出不良的预后作用。 (1)非小细胞肺癌向小细胞肺癌(SCLC)的组型转变。研究报道37例对EGFR-TKI耐药的NSCLC肿瘤组织中有5例患者出现了NSCLC向SCLC组织学类型的转变,而这一转变的分子学机制尚不清楚,Arcila等研究报道了106例非小细胞患者中有3例患者出现了非小细胞肺癌表型向小细胞表型的转变。EGFR-TKI获得性耐药的NSCLC患者的肿瘤标本中检测出NSCLC转化成SCLC其概率约为3%。有研究结果显示其可能的机制是Rb1和TrP53/ target=_blank class=infotextkey>P53在组型转变的过程中起作用。这一研究报道可能会指导下一步治疗。 (2)上皮、间质的表型转化。上皮组织转向间质组(EMT)使肿瘤细胞失去了上皮表型,转化为间质表型。研究报道,EMT的重要特征包括E-钙粘蛋白表达的丧失和非上皮钙粘蛋白(例如N-钙粘蛋白)的表达增加,从而增强了肿瘤细胞的侵袭性,有助于肿瘤细胞的生长和存活。EMT由外部信号启动,例如:HGF,EGFR,转化生长因子(TGF)-b和成纤维细胞生长因子(bFGF)。除了由膜受体触发的这些信号传导途径外,最近的研究还强调了非编码RNA通过控制EMT诱导剂在上皮表型调节中的重要性。已经发现miR-200家族可通过下调Zeb因子的表达来控制EMT。此外,长非编码RNA(lncRNA)MALAT-1通过调节ZEB1,ZEB2和Slug表达并激活Wnt信号传导,促进了EMT转化,使上皮细胞丧失连接蛋白(如E-钙粘蛋白),进而产生了间质标记物,但这一机制尚未完全阐明。 2.2.4 诱导血管生成 NSCLC能够通过不同途径分泌一些血管内皮生长因子及bFGF使得肿瘤间质血管生成促进肿瘤转移导致NSCLC患者预后不良。VEGF通路已成为肺癌治疗的重要靶点,长期以来,VEGF作为一种强有力的促血管生成因子在多种类型的肿瘤中均有表达,研究显示EGFR通路的部分下游因子如MAPK、PI3K可以调控VEGF高表达导致肿瘤的进展及转移使得EGFR-TKI获得性耐药,对于同时兼有EGFR、VEGF突变的NSCLC患者联合应用厄洛替尼和贝伐单抗能够得到额外的临床收益。 2.2.5 其他通路的改变 如:PI3K/AKT、ERK/MAPK、PTEN是EGFR的下游信号分子,而PTEN可降低PI3K/AKT信号传导通路活性,若PTEN缺陷或失活或AKT蛋白过表达,使PI3K/AKT信号通路激活。研究表明,非小细胞肺癌细胞往往存在PI3K/AKT或ERK/MAPK信号通道的激活。另外,研究报道PI3K的p-110α突变可激活PI3K/AKT通路,促使EGFR突变的NSCLC细胞对吉非替尼的IC50值升高减少其凋亡率。研究还发现了敏感非小细胞肺癌细胞株持续表达AKT就可导致耐药产生。研究报道PI3K活化的原因有:(1)相关基因的扩增;(2)mN(能抑制PI3K通路的磷脂酶)的失活丢失或丢失;(3)下游效应蛋白的过度表达(如:AKT蛋白)等,PI3K的活化在EGFR耐药的发展和维持中起到关键作用。 本文综述了关于肺腺癌对第一代EGFR-TKI的多种耐药机制,然而仍有一些耐药机制没有完全展现出来,如是否与肿瘤微环境变化及其他突变有关,尚缺乏对非小细胞肺癌EGFR-TKI耐药后治疗的研究,对如何指导EGFR-TKI耐药后的治疗缺乏认知。EGFR-TKI已经成为晚期EGFR突变的NSCLC患者的标准一线治疗方案,尽管这些患者中大部分癌细胞都存在“敏感的”EGFR突变,并且治疗后大多数患者都有肿瘤缩小的变化,但完全缓解的情况很少,所有患者治疗一段时间后均进展,这表明大量癌细胞不可避免地获得了耐药性,因此对不同机制耐药后治疗问题仍有待进一步解决。

01 /第一代EGFR-TKI药物
02 / EGFR-TKI耐药的分子机制
2.1原发性耐药
2.2获得性耐药